---
title: compare ratxcan with and without loco
date: 2024-08-29
author: Haky Im
editor_options:
chunk_output_type: console
description: compare ratxcan mixed effects with and without loco
---
## load libraries and functions
```{r load libraries and functions}
#options(error=recover)
#options(error=browser)
options(error=NULL)
## compare observed correlation with null correlation
suppressMessages(devtools::source_gist("a925fea01b365a8c605e")) ## load qqR fn https://gist.github.com/hakyim/a925fea01b365a8c605e
suppressMessages(devtools::source_gist("38431b74c6c0bf90c12f")) ## qqunif https://gist.github.com/hakyim/38431b74c6c0bf90c12f
suppressMessages(devtools::source_gist("115403f16bec0a0e871f3616d552ce9b")) ## source ratxcan functions https://gist.github.com/hakyim/115403f16bec0a0e871f3616d552ce9b
suppressMessages(library(tidyverse))
suppressMessages(library(glue))
suppressMessages(library(RSQLite))
#suppressMessages(library(expm))
#suppressMessages(library(readxl))
# install.packages("devtools")
# library("devtools")
# install_github("jdstorey/qvalue")
suppressMessages(library(qvalue))
# if (!require("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#
# BiocManager::install("biomaRt")
##suppressMessages(library(biomaRt))
##install.packages("ggrepel")
suppressMessages(library(ggrepel))
recalc=FALSE
```
## define data and software paths for R
```{r define data and software paths}
WEBDATA="/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data"
PLINK="/Users/haekyungim/bin/plink_mac_20231211/plink"
GCTA="/Users/haekyungim/bin/gcta-1.94.2-MacOS-ARM-x86_64/gcta64"
INPUT <- glue("{WEBDATA}/ratxcan-tutorial") ## this has the input data
OUTPUT <- glue("{WEBDATA}/2023-11-27-ratxcan-tutorial/scratch") ## this has the output data, intermediate results
GENO = glue("{WEBDATA}/2023-11-27-ratxcan-tutorial/data/genotype/")
OUT_TEMPO = glue("{OUTPUT}/testing-2024-08-29")
```
## define data and software for the terminal
```{bash define data and software paths bash}
#| eval: FALSE
WEBDATA="/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data"
PLINK="/Users/haekyungim/bin/plink_mac_20231211/plink"
GCTA="/Users/haekyungim/bin/gcta-1.94.2-MacOS-ARM-x86_64/gcta64"
INPUT=$WEBDATA/ratxcan-tutorial
OUTPUT=$WEBDATA/2023-11-27-ratxcan-tutorial/scratch
GENO=$WEBDATA/2023-11-27-ratxcan-tutorial/data/genotype/
OUT_TEMPO=$OUTPUT/testing-2024-08-29
```
## read grm matrix
```{r read GRM mat}
grm_mat <- read_GRMBin(glue("{OUTPUT}/rat6k_autosome.grm"))
```
## define myplot
```{r define myplot}
myplot <- function(tempres, post_titulo="",semilla="") {
# Create a data frame with specific columns
df <- data.frame(
p0.01_yes = apply(tempres$pmat_correct, 2, function(x) mean(x < 0.01)),
p0.01_no = apply(tempres$pmat_raw, 2, function(x) mean(x < 0.01)),
p0.05_yes = apply(tempres$pmat_correct, 2, function(x) mean(x < 0.05)),
p0.05_no = apply(tempres$pmat_raw, 2, function(x) mean(x < 0.05)),
p0.10_yes = apply(tempres$pmat_correct, 2, function(x) mean(x < 0.10)),
p0.10_no = apply(tempres$pmat_raw, 2, function(x) mean(x < 0.10))
# ... [rest of your code for creating df] ...
)
# Pivot the data frame to long format, specifying the columns to keep
df_long <- pivot_longer(df, cols = starts_with("p"))
df_long <- df_long %>% separate(name,into = c("threshold","corrected"),sep="_") %>% rename(proportion=value)
# Rename the name column to replace p0.xx with p<0.xx
df_long <- df_long %>%
mutate(threshold = gsub("p0\\.", "p<0.", threshold))
# Create boxplots with mean
pp <- ggplot(df_long, aes(x = threshold, y = proportion, fill = corrected)) +
geom_boxplot(alpha = 0.6) +
stat_summary(fun = mean, geom = "point", shape = 3, size = 2, stroke = 2, color = "blue", #position = position_dodge(width = 0.8)) +
position = position_dodge(width = -0.1)) +
#stat_summary(fun = mean, geom = "crossbar", size = .5, color = "blue") +
#stat_summary(fun = mean, geom = "crossbar", size = .5, color = "darkgray") +
geom_hline(yintercept = c(0.01, 0.05, 0.10), linetype = "dashed", color = "gray") +
theme_minimal(base_size = 17) +
ggtitle(glue("Type I Error Calibration {semilla} {post_titulo}")) +
xlab("significance") + ylab("false positive rate")
pp
}
```
### NEED DEBUG simulate $Y = Sigma^{1/2}\epsilon$ and run assoc with expr_mat
```{r LONG RUN Y sigma^.5 epsimat}
#| eval: FALSE
nsam=nrow(grm_mat)
#ind=1:nsam
ind=1:1000
test_mat = grm_mat[ind,ind]
nsam=nrow(test_mat)
Sigma = test_mat * h2 + (1 - h2) * diag(rep(1,nsam))
Sig_eigen = eigen(Sigma)
rownames(Sig_eigen$vectors) = rownames(Sigma)
##sighalf = Sig_eigen$vectors %*% diag( sqrt( Sig_eigen$values ) ) %*% t(Sig_eigen$vectors)
## make this multiplication more efficient using sweep
sighalf = Sig_eigen$vectors %*% sweep(t(Sig_eigen$vectors),1,sqrt( Sig_eigen$values ),"*")
sim_sigma_pheno = sighalf %*% matrix(rnorm(nsam * nsim), nsam, nsim)
sim_sigma_pheno=cbind(FID=rownames(sim_sigma_pheno),IID=rownames(sim_sigma_pheno),as.data.frame(sim_sigma_pheno))
tic=Sys.time()
tempres_sigma_pheno <- lmmGRM(sim_sigma_pheno,grm_mat, h2,pred_expr,pheno_id_col=1, pheno_value_cols=2+(1:nsim))
toc=Sys.time()
toc - tic
pp<-myplot(tempres_sigma_pheno,post_titulo = glue("sigma_pheno n= {nsam} - ii={ii}"))
cat(ii,"\n")
print(pp)
```
## read gene annotation
```{r read gene annotation}
#gene_annotation <- readRDS(glue("{INPUT}/data/expression/gene_annotation.RDS"))
gene_annotation <- readRDS(glue("{WEBDATA}/2023-11-27-ratxcan-tutorial/data/expression/gene_annotation.RDS"))
```
## read predicted expression
```{r read predicted expression}
read_pred_expr = function(filename)
{
##usage: Br_pred_expr = read_pred_expr(glue("{OUTPUT}/Br-hki-rat6k__predict.txt"))
pred_expr <- vroom::vroom(filename) %>%
select(-FID) %>% # Remove the FID column
mutate(IID = str_split(IID, "_", simplify = TRUE)[, 1]) # Keep the first part of IID
pred_expr
}
# pred_expr = read_pred_expr(glue("{OUTPUT}/Br-hki-rat6k__predict.txt"))
# ## WHYYYY br-hki-rat6k__predict.txt has no chr 1????
# df1= tibble(gene=names(pred_expr)) %>% mutate(isinac=TRUE) %>% left_join(gene_annotation,by=c("gene"="gene_id"))
# df1 %>% count(chr)
# cat("{OUTPUT}/Br_rat6k__predict.txt has no chr 1 genes!!! Where did I lose them?\n")
pred_expr = read_pred_expr(glue("{OUTPUT}/AC-filtered__predict.txt"))
## checked that I have chr1 genes
df1= tibble(gene=names(pred_expr)[-1]) %>% mutate(isinac=TRUE) %>% left_join(gene_annotation,by=c("gene"="gene_id"))
df1 %>% count(chr)
cat("{OUTPUT}/AC-filtered__predict.txt has genes in ",length(unique(df1$chr)), "chr")
```
## define function lmm with GRM
```{r define lmmGRM function}
## HERE WE USE THE FULL GRM MATRIX AND CALCULATE THE INVERSE OF THE SIGMA MATRIX
## define lmm association function
lmmGRM = function(pheno, grm_mat, h2, pred_expr,pheno_id_col=1,pheno_value_cols=6:6,out=NULL)
{
## input pheno is a data frame with id column pheno_id_col=1 by default
## phenotype values are in pheno_value_cols, 6:6 by default (SCORE column location in plink output), it can have more than one phenotype
## but h2 has to be the same, this is useful when running simulations with different h2
## call lmmXcan(pheno %>% select(IID,SCORE))
## format pheno to matrix form
phenomat <- as.matrix(pheno[,pheno_value_cols])
rownames(phenomat) <- pheno[[pheno_id_col]]
## turn pred_expr into matrix with rownames =IID, keep only IIDs in ymat
exp_mat = as.matrix(pred_expr %>% select(-IID))
rownames(exp_mat) = pred_expr$IID
## align pheno and expr matrices
idlist = intersect(rownames(phenomat), rownames(exp_mat))
nsam = length(idlist)
## CALCULATE SIGMA
ID_mat = diag(rep(1,nsam))
#testing_scale_grm = TRUE
#if(testing_scale_grm) grm_mat = sweep( sweep(grm_mat,2, 1/sqrt(diag(grm_mat)),"*"), 1, 1/sqrt(diag(grm_mat)),"*")
Sigma = grm_mat[idlist,idlist] * h2 + (1 - h2) * ID_mat
Sig_eigen = eigen(Sigma)
rownames(Sig_eigen$vectors) = rownames(Sigma)
isighalf = Sig_eigen$vectors %*% diag( 1 / sqrt( Sig_eigen$values ) ) %*% t(Sig_eigen$vectors)
## perform raw association
cormat_raw = matrix_lm(phenomat[idlist,, drop = FALSE], exp_mat[idlist,])
pmat_raw = cor2pval(cormat_raw,nsam)
colnames(pmat_raw) <- gsub("cor_", "pval_", colnames(pmat_raw))
## perform corrected association
cormat_correct = matrix_lm(isighalf%*% phenomat[idlist,, drop = FALSE], isighalf %*% exp_mat[idlist,])
pmat_correct = cor2pval(cormat_correct,nsam)
colnames(pmat_correct) <- gsub("cor_", "pval_", colnames(pmat_correct))
if(!is.null(out))
{
saveRDS(cormat_correct,file = glue("{out}_cormat_correct.RDS"))
saveRDS(pmat_correct, file = glue("{out}_pmat_correct.RDS"))
saveRDS(cormat_raw, file = glue("{out}_cormat_raw.RDS"))
saveRDS(pmat_raw, file = glue("{out}_pmat_raw.RDS"))
}
res = list(
cormat_correct=cormat_correct,
pmat_correct=pmat_correct,
cormat_raw=cormat_raw,
pmat_raw=pmat_raw)
res
}
```
## run ratXcan
```{r}
## read simulated Y
pheno = read_table(glue("{WEBDATA}/2023-11-27-ratxcan-tutorial/scratch/sim/tempo/PRS_output_100-32240.profile"))
## calculate h2 of pheno
## mpheno 4 means fourth column after FID and IID
## $GCTA --grm $OUTPUT/rat6k_autosome --reml --pheno $OUTPUT/sim/tempo/PRS_output_100-32240.profile --mpheno 4 --out $OUTPUT/test-2024-08-29
## create phenotype with h2
h2=0.5
pheno$trait = (pheno$SCORE - mean(pheno$SCORE))/sd(pheno$SCORE) * sqrt(h2) + sqrt(1 - h2)*rnorm(nrow(pheno))
if(recalc)
{res = lmmGRM(pheno, grm_mat, h2, pred_expr,pheno_id_col=1,pheno_value_cols=7:7,out=NULL)
saveRDS(res,glue("{OUT_TEMPO}/ratxcan-ac-res.RDS"))
} else
res = readRDS(glue("{OUT_TEMPO}/ratxcan-ac-res.RDS"))
```
## calc GRM without chr 1
```{bash calculate GRM without chr01}
#| eval: false
$PLINK --bfile $GENO/rat6k_autosome --chr 1 --write-snplist --out $OUT_TEMPO/chr1_snps
$GCTA --bfile $GENO/rat6k_autosome --exclude $OUT_TEMPO/chr1_snps.snplist --make-grm-bin --out $OUT_TEMPO/rat6k_autosome_loco_chr01 --thread-num 8
## that took
```
```{text }
*******************************************************************
* Genome-wide Complex Trait Analysis (GCTA)
* version v1.94.1 Mac
* (C) 2010-present, Yang Lab, Westlake University
* Please report bugs to Jian Yang <jian.yang@westlake.edu.cn>
*******************************************************************
Analysis started at 16:52:27 CDT on Thu Aug 29 2024.
Hostname: MED-ML-464.local
Accepted options:
--bfile /Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome
--exclude /Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/testing-2024-08-29/chr1_snps.snplist
--make-grm-bin
--out /Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/rat6k_autosome_loco_chr1
--thread-num 8
Note: the program will be running on 8 threads.
Reading PLINK FAM file from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.fam].
5628 individuals to be included from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.fam].
Reading PLINK BIM file from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.bim].
179895 SNPs to be included from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.bim].
Reading a list of SNPs from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/testing-2024-08-29/chr1_snps.snplist].
17602 SNPs are excluded from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/testing-2024-08-29/chr1_snps.snplist] and there are 162293 SNPs remaining.
Reading PLINK BED file from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.bed] in SNP-major format ...
Genotype data for 5628 individuals and 162293 SNPs to be included from [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/data/genotype//rat6k_autosome.bed].
Calculating allele frequencies ...
Recoding genotypes (individual major mode) ...
Calculating the genetic relationship matrix (GRM) ... (Note: default speed-optimized mode, may use huge RAM)
Summary of the GRM:
Mean of diagonals = 0.990807
Variance of diagonals = 0.00404564
Mean of off-diagonals = -0.000176097
Variance of off-diagonals = 0.00216189
GRM of 5628 individuals has been saved in the file [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/rat6k_autosome_loco_chr1.grm.bin] (in binary format).
Number of SNPs to calculate the genetic relationship between each pair of individuals has been saved in the file [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/rat6k_autosome_loco_chr1.grm.N.bin] (in binary format).
IDs for the GRM file [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/rat6k_autosome_loco_chr1.grm.bin] have been saved in the file [/Users/haekyungim/Library/CloudStorage/Box-Box/LargeFiles/imlab-data/data-Github/web-data/2023-11-27-ratxcan-tutorial/scratch/rat6k_autosome_loco_chr1.grm.id].
```
## compute GRM loco chr01 and lmmGRM with updated h2
```{r compute GRM no chr01 and lmmGRM}
grm_mat_no_chr01 = read_GRMBin(glue("{OUT_TEMPO}/rat6k_autosome_loco_chr01.grm"))
## need to recalculate h2
# $GCTA --reml --grm $OUT_TEMPO/rat6k_autosome --out $OUT_TEMPO/rat6k_autosome_loco_chr01 --thread-num 8
## save the phenotype with error added
write_tsv(pheno %>% select(IID, FID, trait),file=glue("{OUT_TEMPO}/pheno_PRS_output_100-32240.txt"))
# $GCTA --grm $OUT_TEMPO/rat6k_autosome_loco_chr01 --reml --pheno $OUT_TEMPO/pheno_PRS_output_100-32240.txt --mpheno 1 --out $OUT_TEMPO/loco_chr01 --thread-num 8
h2_loco_chr01 = read_tsv(glue("{OUT_TEMPO}/loco_chr01.hsq")) %>% filter(Source=="V(G)/Vp") %>% pull(Variance)
if(recalc)
{
res_no_chr01 = lmmGRM(pheno, grm_mat_no_chr01, h2= h2_loco_chr01, pred_expr,pheno_id_col=1,pheno_value_cols=7:7,out=NULL)
saveRDS(res_no_chr01,file=glue("{OUT_TEMPO}/ratxcan-ac-res-loco-chr01.RDS"))
} else res_no_chr01=readRDS(glue("{OUT_TEMPO}/ratxcan-ac-res-loco-chr01.RDS"))
```
## compare pvalues in chr 1 vs other chromosomes
```{r compare pvalues in chr 1 vs other chromosomes}
pmat2df = function(pmat)
{
df = data.frame(pmat)
df$gene = rownames(pmat)
rownames(df) = NULL
df %>% left_join(gene_annotation %>% select(gene_id,chr,gene_name),by=c("gene"="gene_id") )
}
df_loco_chr01 = pmat2df(res_no_chr01$pmat_correct)
df = pmat2df(res$pmat_correct)
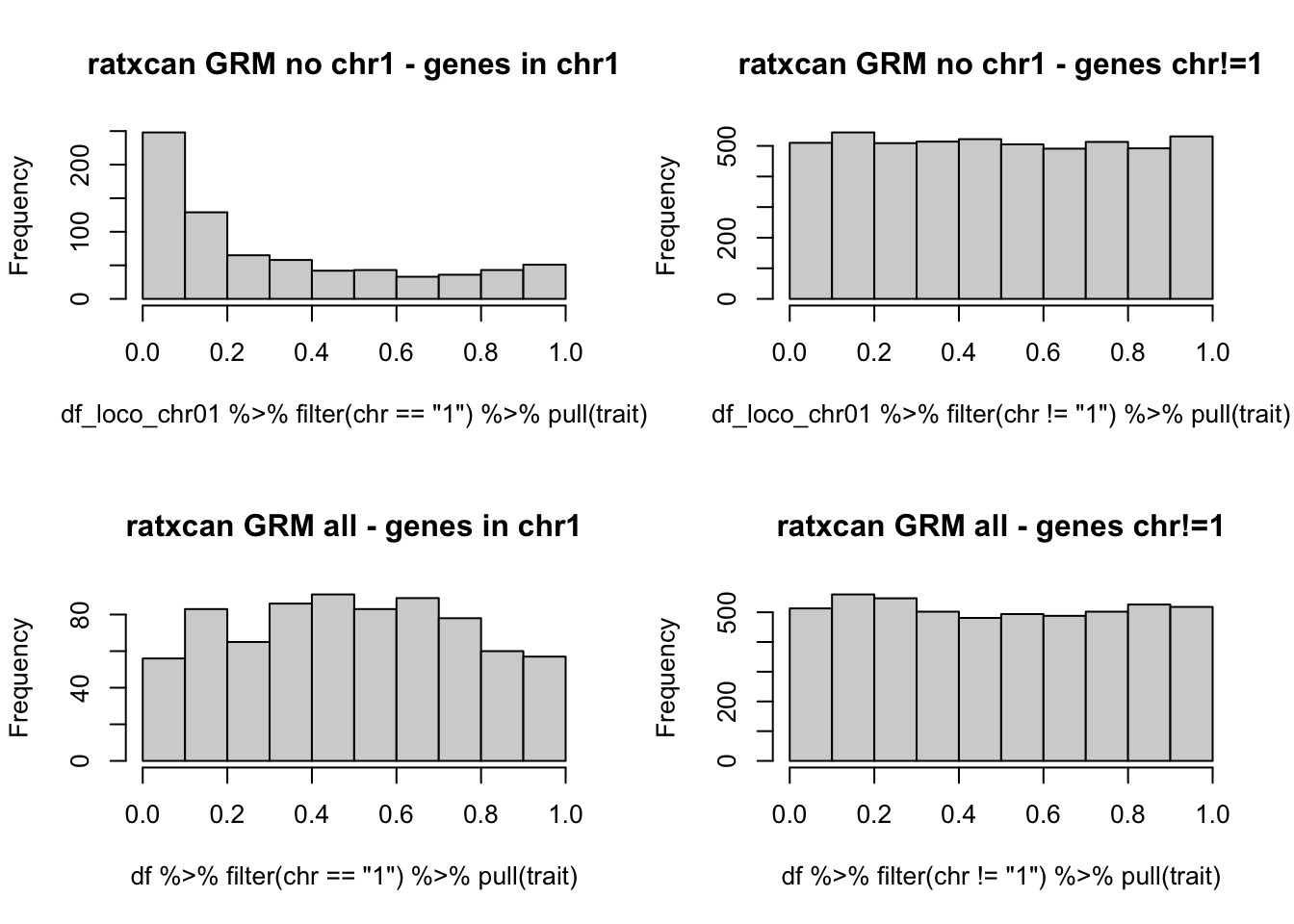
par(mfrow=c(2,2))
hist(df_loco_chr01 %>% filter(chr=="1") %>% pull(trait),main="ratxcan GRM no chr1 - genes in chr1")
hist(df_loco_chr01 %>% filter(chr!="1") %>% pull(trait),main="ratxcan GRM no chr1 - genes chr!=1")
hist(df %>% filter(chr=="1") %>% pull(trait),main="ratxcan GRM all - genes in chr1")
hist(df %>% filter(chr!="1") %>% pull(trait),main="ratxcan GRM all - genes chr!=1")
par(mfrow=c(1,1))
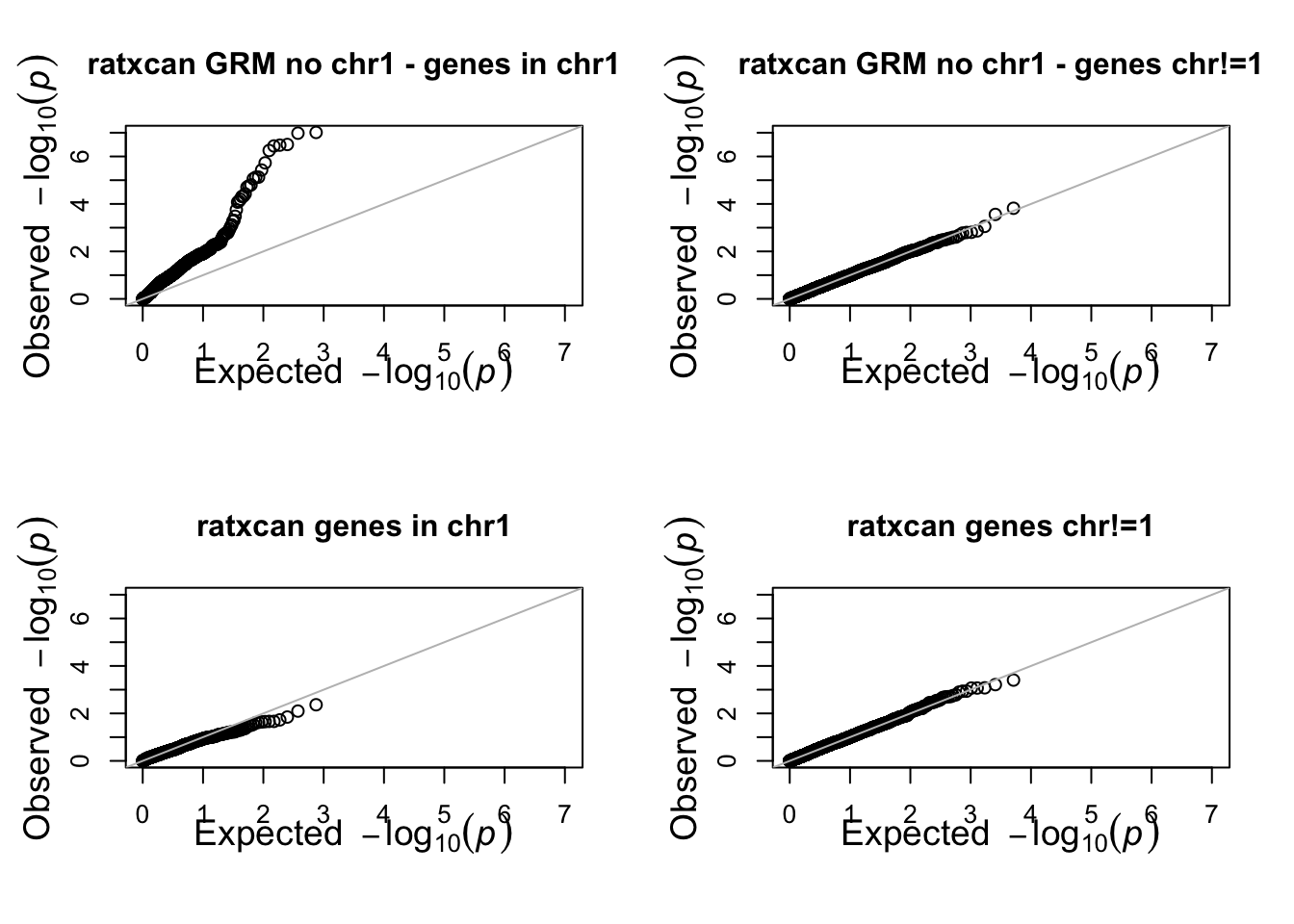
par(mfrow=c(2,2))
rango = range(-log10(c(df_loco_chr01$trait, df$trait)))
qqunif(df_loco_chr01 %>% filter(chr=="1") %>% pull(trait),main="ratxcan GRM no chr1 - genes in chr1",BH=FALSE,CI=FALSE,xlim=rango,ylim=rango)
qqunif(df_loco_chr01 %>% filter(chr!="1") %>% pull(trait),main="ratxcan GRM no chr1 - genes chr!=1",BH=FALSE,CI=FALSE,xlim=rango,ylim=rango)
qqunif(df %>% filter(chr=="1") %>% pull(trait),main="ratxcan genes in chr1",BH=FALSE,CI=FALSE,xlim=rango,ylim=rango)
qqunif(df %>% filter(chr!="1") %>% pull(trait),main="ratxcan genes chr!=1",BH=FALSE,CI=FALSE,xlim=rango,ylim=rango)
par(mfrow=c(1,1))
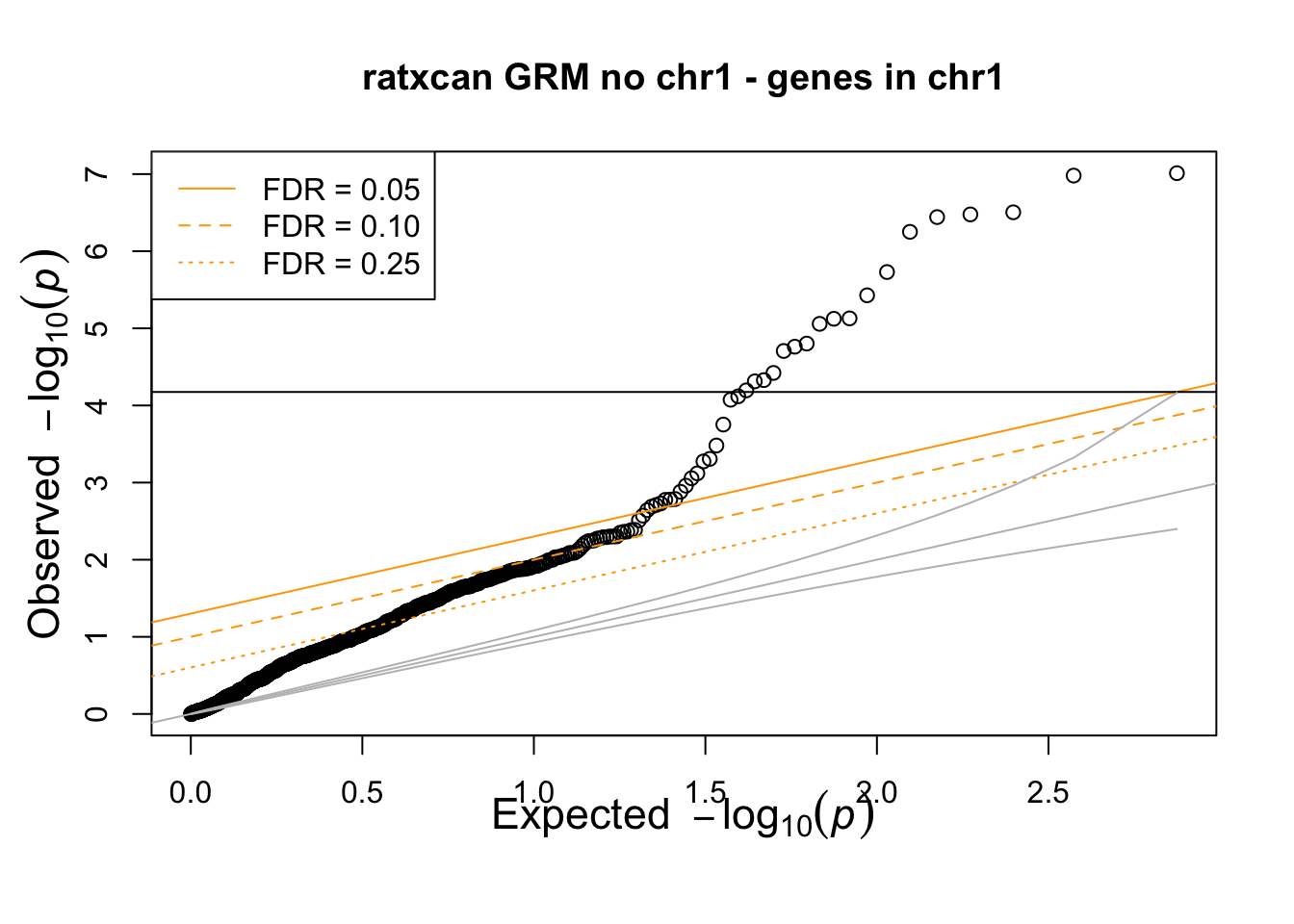
qqunif(df_loco_chr01 %>% filter(chr=="1") %>% pull(trait),main="ratxcan GRM no chr1 - genes in chr1")
```
::: {.callout-warning}
## Take home
removing chr1 from GRM negates the correction of ratxcan in chr1.
:::
> phenotype was simulated to have a relatedness modeling random effect (covariance=GRM), no conexion to any of the genes. Any significant association should be considered false positive.